

Compound signatures are comprised of clusters, which are in turn comprised of groups of peaks. Compound Builder provides a variety of tools for creating and manipulating these signatures.
To ensure compatibility with both the default Chenomx Reference Library as well as your experiment spectra, acquire your new Compound Signature using careful Sample Preparation and NMR acquisition parameters that will correspond to your Experiment Spectra.
 |
Sample Preparation Steps
-
Prepare a stock solution with a known concentration of your compound
-
Prepare an Internal Standard Solution using either DSS or TSP with 100% D2O
-
Mix Stock Solution with Internal Standard Solution at 9:1 ratio as NMR Instrument requires 10% deuterium to lock.
-
Shake or vortex mixture and adjust pH to 7.0
-
Acquire spectrum by NMR using recommended parameters:
-
Temperature = 25 °C
-
Acquisition time = 4 s
-
Relaxation delay = 1 s
-
Spectral width = 12 ppm
-
The FID folder acquired from the NMR Instrument can be opened with the Chenomx Processor to create a CNX file. This CNX file can be opened in Compound Builder to create your new Compound Reference Spectrum file (*.XCPD)
After acquiring your FID file from the NMR Instrument, open and process your Raw Spectrum in the Processor and create a CNX file for compatibility with the Compound Builder. Refer to “Processing Steps” for best practices.
-
Open your processed CNX file reflecting the isolated signals of your new compound.
-
Ensure NMR field strength used to obtain the spectrum matches with the Compound Details Menu.
-
Enter Concentration of compound as it was measured in NMR tube after adding internal standard.
-
Enter Compound Name; pH; Pulse Sequences; UUID as required
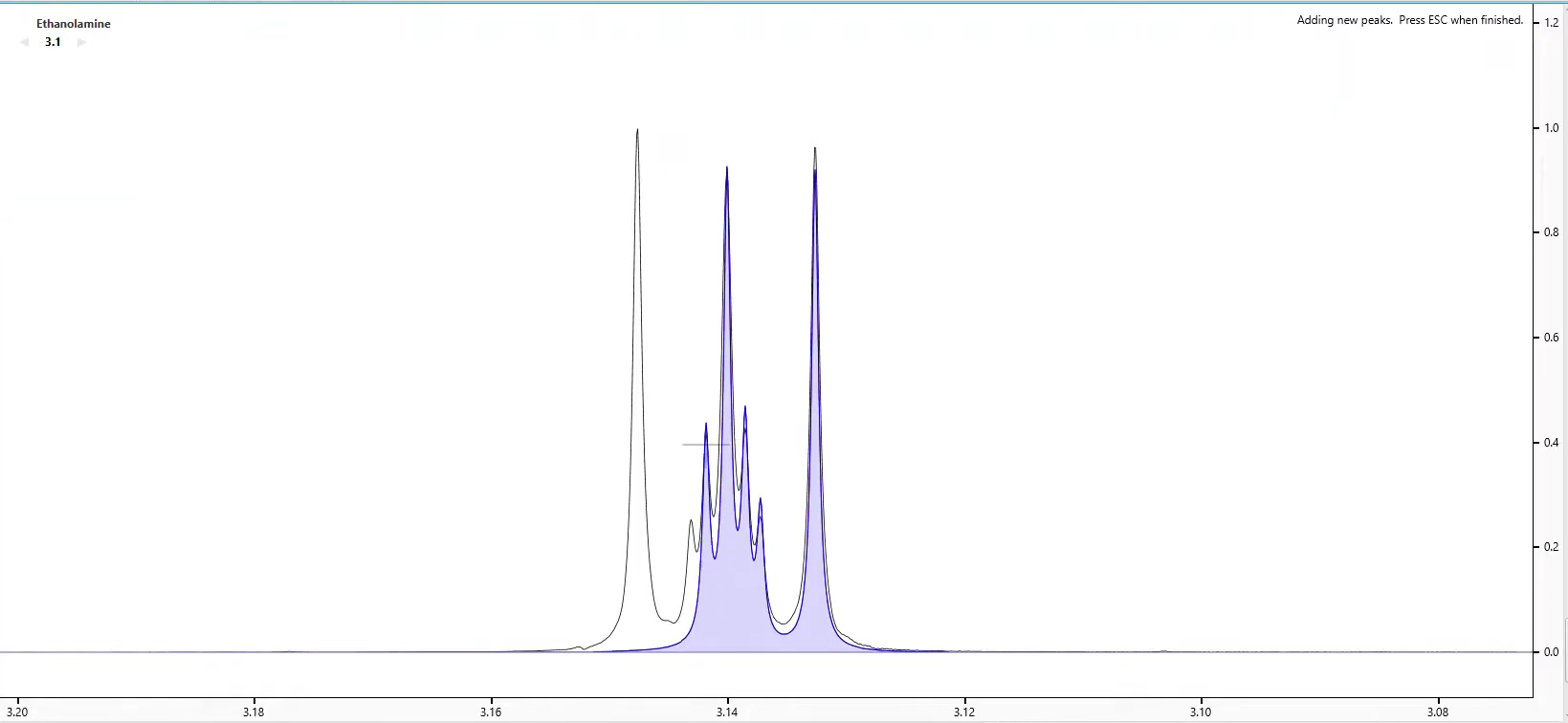
Begin Adding Peaks, Defining Clusters and Optimizing Shapes to create your Compound Reference Library Spectrum.
 |
You can add, select, and delete peaks or clusters by clicking on them in the graph, or in the Information Panel.
| Menu Location | Compound > Add Peaks |
| Icons |
|
Add peaks to the current signature by clicking on the Compound Spectrum.
-
Switch to Add Peaks mode and the cursor will change to a large crosshair.
-
Click the crosshair within the first Compound Spectrum Cluster to create your first peak. Or... Right-click anywhere in the Spectrum Graph that does not contain a peak and select Add Peak at x.xx ppm. The click point defines both the position and the height of the new peak. You are free to change both after you have added the peak.
-
To add additional peaks to the Compound Selected Cluster, hold the Ctrl or Cmd key while clicking for new peaks.
-
Continue to add peaks until all peaks within the Compound Spectrum have been accounted for. When finished, click Compound > Stop Adding Peaks.
-
NOTE: The new peak appears in the peak list in numerical order with the currently displayed peaks, not necessarily at the beginning or end of the list.
Selecting, Deleting Peaks and Clusters
-
To Select a Cluster: ensure 'Selection by Cluster' is turned on, then click on the cluster in the Spectrum Graph, or; Click on the Cluster's position in the Information Panel, or; Double Click on the Cluster.
-
To Select a Peak: Ensure 'Selection by Cluster' is turned off, then click on the cluster in the Spectrum Graph, or; Click on the Peak's position in the Information Panel.
-
To Select Multiple Peaks or Clusters: turn 'Selection by Cluster' on or off, as needed; Hold down the Ctrl or Cmd key, then click several peaks or clusters, either in the Spectrum Graph, the Information Panel, or both. You can hold down Shift instead to select a range of peaks or clusters.
-
To Delete Peaks or Clusters: Select the peaks or clusters; Click 'Delete Selected Peaks'.
Additional Notes
-
When a cluster is selected, only the peaks associated with that cluster are available to select via the Information Panel.
-
When no clusters are selected, no peaks are visible or available to select via the Information Panel.
-
If a peak is not visible in the Information Panel, select a different cluster.
-
Delete multiple peaks or clusters by selecting them via the Spectrum Graph or the Information Panel.
-
Right-click on a selection in the Spectrum Graph or Information Panel and click Delete Selected Peaks.
| Menu Location | Compound > Delete Selected Peaks |
| Icon |
|
In most cases, the initial properties of a peak do not accurately match the spectrum overlay. Modify the height or frequency (center position) of one or more peaks to arrive at a signature that fits the spectrum overlay shape.
| Menu Location | Compound > Edit Selected Peaks... |
| Icon |
|
Set peak properties to specific values
-
Double-click on a peak or cluster in the Information Panel, Select the peaks or clusters that require adjustment and open the 'Edit Selected Peaks' dialog, or; right-click on a selection in the Spectrum Graph or Information Panel and select Edit Selected Peaks NOTE: When you enter specific amplitudes and widths, all of the selected peaks are set to the specified values, regardless of which clusters they are associated with.
-
Enter the desired center (position in ppm), amplitude (height in si) and width (in sw) to apply to all selected peaks or clusters and click OK.
Adjust peak properties
Select the peaks or clusters that you would like to adjust and do any of the following:
-
Click and drag the selected peaks or clusters in the Spectrum Graph to adjust their positions and heights. Use the blue triangles on the horizontal and vertical axes for fine control over position and height, respectively.
-
Click and drag the blue triangles on either side of the selected peaks and clusters, at approximately half-height, to adjust their widths.
-
Use the W and S keys or the up and down arrow keys to adjust the height of the selection up and down; The A and D keys or the left and right arrow keys to adjust left and right; and the Q and E keys to display the next or previous cluster.
Tips and Tricks
-
When you adjust the properties for a single peak, you can use mathematical functions of 'ph' instead of constant values to set the center, height and width of the peak. To insert an function expression instead of a single value, right-click in the text box that you would like to change, then click Expression....
-
To view or edit an existing expression, click the expression icon (
 ) in the text box that you would like to change.
) in the text box that you would like to change.
Especially when building more complex compounds, it may be necessary to group peaks from one or more separate clusters into a single cluster of their own. Grouping peaks into clusters ensures that all of the selected peaks move together when you use the compound signature for profiling, and that they obey the same transform window settings.
| Menu Location | Compound > Group Selected Peaks as Cluster |
| Icon |
|
How to group peaks as a cluster
-
Right-click on a selection in the Spectrum Graph or Information Panel and select Group Selected Peaks as Cluster to group the selected peaks directly.
-
After grouping peaks into a new cluster, you may want to use the Edit Selected Peaks dialog to set them all to a common width.
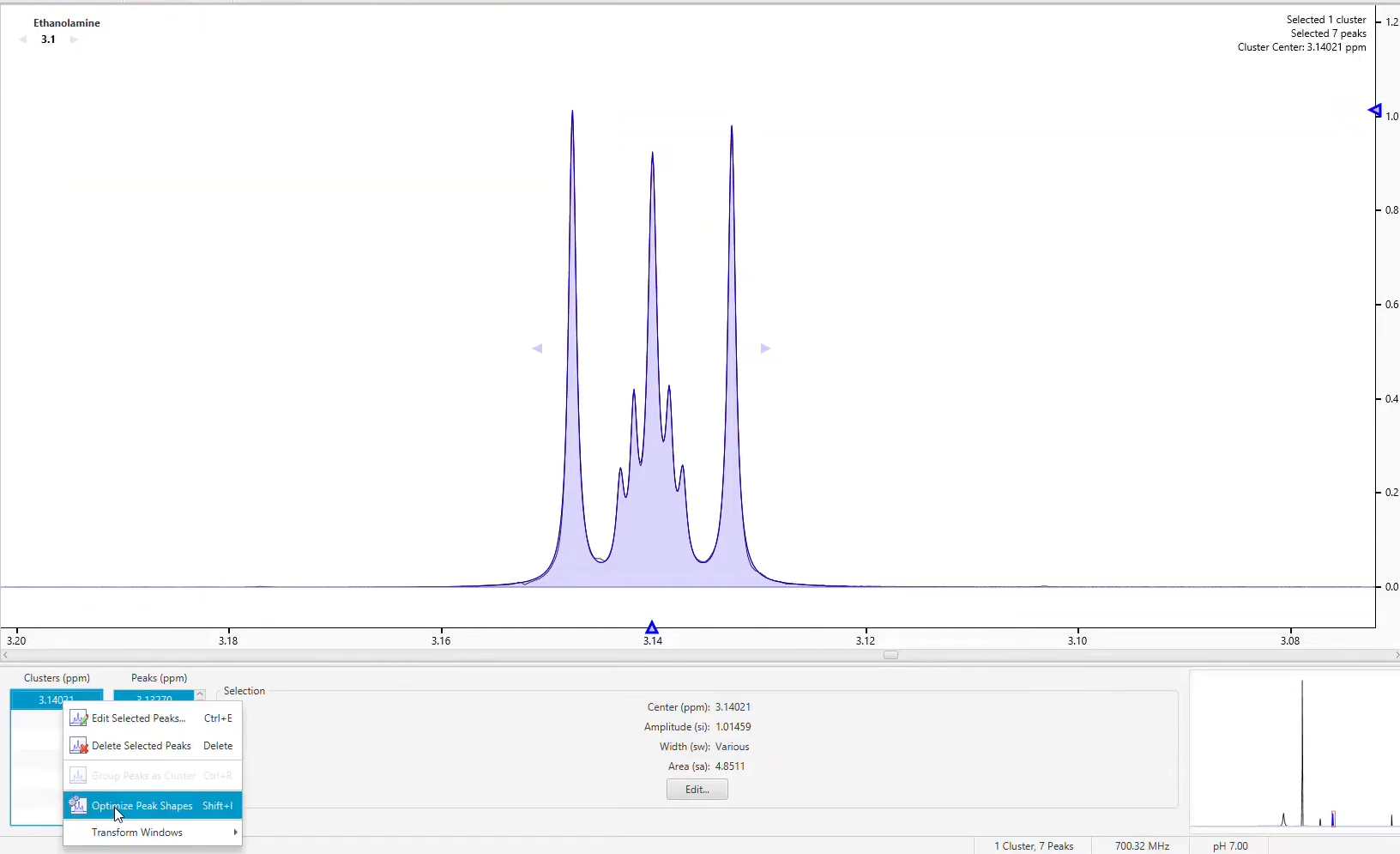
Adding peaks to a signature and adjusting them to approximately model a particular region of the spectrum is fairly simple. However, the task of fine-tuning those peaks to accurately match the lineshape in that region can be tedious. Once you have approximated the shape of a region of the spectrum, use Compound Builder's optimization algorithms to rapidly obtain a more refined match to that region.
| Menu Location | Tools > Optimize Selected Peak Shapes |
| Icon |
|
Optimize the shape of peaks in a Compound Signature
-
Optimization will generally give better results if your starting shape is reasonably close to the actual shape of the spectrum. In particular, it is important to have the right number of peaks. Simulating the compound in Spin Simulator can often give you a very good starting shape.
-
Select the peaks and clusters (fewer than 50) that you would like to optimize and Click 'Optimize Selected Peak Shapes'.
 |
Establishing transform windows for your compounds limits the amount that individual clusters can move within the Profiler module and improves the quality of your profiles.
If you set transform windows manually, ensure that they are wide enough to account for the degree of variability in cluster position that you expect to see in your samples. For example, if you routinely analyze spectra with widely varying pH, you will need larger transform windows than if your sample spectra are always close to the same pH.
The most common influences on the position of a cluster in a compound are pH and ion concentrations. To manually set accurate transform windows for a compound, consider acquiring spectra of the pure compound at the outer limits of the conditions you expect to encounter when profiling the compounds in experimental mixtures. This makes it easier to determine maximum extent of these effects on the cluster positions in your compounds.
In the absence of specific information about the behavior of a cluster, you can set a default transform window for the cluster. The default transform window is 0.04 ppm wide, centered on the current cluster center. For a cluster appearing at 1.000 ppm, the default transform window sets lower and upper limits of 0.98 and 1.02 ppm, respectively.
| Menu Location | Compound > Transform Window > Edit for Selected Cluster... |
| Icon |
|
How to set the transform window for a cluster
-
Select the cluster for which you would like to set a transform window and open the 'Edit Transform Window' Dialog.
-
Select Limit transforms to, and enter the new lower and upper limits for the selected cluster. Click OK
How to change the Transform Window for a cluster
-
Select the cluster whose transform window you would like to change and open the 'Edit Transform Window' dialog.
-
Enter the new lower and upper limits for the selected cluster and click OK.
| Menu Location | Compound > Transform Window > Set Default on Selected Clusters |
| Icon |
|
| Menu Location | Compound > Transform Window > Set Default on All Clusters |
| Icon |
|
How to set the default Transform Window for cluster(s).
-
Single Cluster: Select the cluster for which you would like to set the transform window and on the Compound Menu, select Transform Window > Set Default on Selected Cluster.
-
All Clusters: On the Compound menu, select Transform Window > Set Default on All Clusters.
| Menu Location | Compound > Transform Window > Clear for Selected Clusters |
| Icon |
|
| Menu Location | Compound > Transform Window > Clear for All Clusters |
| Icon |
|
How to clear the transform window for cluster(s)
-
Single Cluster: Select the cluster for which you would like to clear the transform window and on the Compound menu, select Transform Window > Clear for Selected Clusters.
-
All Clusters: On the Compound menu, select Transform Window > Clear for All Clusters.
| Menu Location | Tools > pH Sensitivities... |
| Icon |
|
How to set pH sensitive Transform Windows
-
On the Tools menu, click pH Sensitivities and Click Load Data Points.
-
Select the signatures that you would like to use to describe pH sensitive centers and transform windows for the compound.
-
Click Set Data Points, then; Click Optimize Lines.
-
Review the clusters in the compound. Click Accept to apply the automatically generated expressions for cluster centers and transform windows to your compound..
Tips and Tricks
-
You should always apply transform windows to any compound that you create. Compounds without transform windows will always appear in the Compound Table in Profiler when performing a Quick Search by region.
-
Many of the operations described above for single clusters (setting default transform windows, clearing transform, etc.) can also be performed on multiple selected clusters at once.
-
A transform window applies to the center of a cluster, not the positions of its outermost peaks. Consider only cluster centers when you manually set transform windows for your compounds.
-
After you have set a transform window for a cluster, if you try to move the cluster beyond either limit of the transform window, the limit moves to accommodate the new position of the cluster. If the transform window was an expression, adjusting it in this manner will replace the expression with a constant.
-
You may need to acquire multiple spectra to determine appropriate transform windows for a compound. Remember that some compounds, and some clusters within a compound, are more sensitive than others to pH changes and other matrix effects.
-
When you create pH sensitive compounds, expressions (functions of 'pH') for the transform windows are calculated automatically, based on the set of spectra that you select. You can view or edit this expressions by clicking on the expression icon (
) in the text box for the transform.
Related Topics